

Project 2: The Role of Dietary Sodium in Blood-Brain Barrier Erosion and Microcrystal Formation in the Amygdala
Authors: Dr. A. Ziggy Semmelwise & Professor Pluto
All authors use protected pseudonyms due to ongoing suppression efforts by industrial stakeholders.Abstract
This longitudinal study investigates the role of synthetic sodium-based food preservatives, particularly NaCl-9, in the progressive erosion of the blood-brain barrier (BBB) and the formation of microcrystalline sodium structures within key limbic structures. Employing histological analysis of brain tissue, controlled rodent exposures, MRI scan data from vulnerable communities, and in vitro endothelial barrier models, we present a cohesive pattern of neurological disruption arising from chronic exposure to legacy sodium compounds. The findings establish a plausible pathophysiological mechanism by which these additives penetrate neural tissue, destabilize emotional regulation, and embed themselves in microanatomical structures with lasting effects. These results call for an immediate re-evaluation of past regulatory approvals and a re-investigation into long-term sodium additive exposure impacts on mental health.
Introduction
Industrial sodium additives, introduced in the late 20th century to enhance food preservation and shelf stability, remain among the least regulated compounds in terms of long-term neurological outcomes. NaCl-9, a particularly resilient synthetic variant, was introduced to commercial and military food systems under minimal oversight, prized for its anti-clumping, anti-bacterial, and thermal properties. While the compound was eventually withdrawn from the GRAS database, its neurobiological legacy has never been fully investigated.
This paper assembles empirical evidence from multiple domains to examine the plausibility of NaCl-9 infiltrating human brain tissue through BBB compromise. By combining controlled exposures, imaging, and postmortem analysis, we aim to address the gap left by institutional silence around long-term sodium exposure in the CNS. The neurotoxic potential of halogenated sodium variants, especially those with bromine stabilizers, raises questions not just about cumulative health risks but also about policy-level inertia and possible systemic negligence in regulatory science.
Methodology
1. Autopsy Mineral Detection
The autopsy investigation was conducted using human tissue obtained under ethical agreements from regional morgues situated in areas of elevated dietary sodium exposure. Our primary concern was identifying inorganic mineral deposits in brain regions typically absent of such features. After cryosectioning at high fidelity, we applied multiple stains in sequence Von Kossa to detect phosphate and calcium deposits, and counterstains to control for lipofuscin and hemosiderin interference. The specimens were fixed with paraformaldehyde and dehydrated via graded ethanol baths before being embedded in resin.
Scanning electron microscopy (SEM) was conducted in partnership with a donated JEOL JSM-5510 system calibrated against known brominated sodium crystals synthesized in our lab. Energy-dispersive X-ray spectroscopy (EDX) was configured to detect elements from sodium (Na) through bromine (Br), with peak analysis algorithms running through custom MATLAB scripts to isolate relevant molar profiles. Blinded analysis was enforced by segmenting specimen IDs from origin counties until final review.
2. Rat Open-Field Trials
We designed a longitudinal animal trial using three cohorts across matched age and weight characteristics. The rats were housed in climate-controlled chambers built from surplus laboratory enclosures, retrofitted to meet AALAS humidity and ventilation guidelines. Behavior was observed and documented using both open-field maze grids (manually mapped) and ultrasonic microphones capturing inaudible frequencies of distress. Behavior scoring followed a modified Boissier scale.
Feeding protocols involved kibble batches synthesized to isolate NaCl-9 as the only divergent variable. These were produced under sterile conditions and subjected to monthly compound verification via liquid chromatography. Monthly euthanasia of a subset per group permitted time-sequenced necropsy under perfusion with Evans Blue and saline. Cryosectioning followed, and neuroanatomical zones were analyzed using histochemical and immunofluorescent markers for GFAP, Iba1, and tight-junction proteins. Statistical analysis employed ANOVA and Tukey post-hoc tests to isolate behavior-to-crystal correlations.
3. MRI Data Mining
MRI data was sourced through decommissioned Department of Public Health imaging arrays, accessed under hardware salvage contracts. Drives recovered contained partial patient data scrubbed of personal identifiers, retaining only age, sex, ZIP code, and scan date. T2-weighted and SWI sequences were exported into NIfTI format and processed with 3D Slicer, ITK-SNAP, and an internal Na-mineral signal isolation plugin. Registration and normalization were conducted against the MNI-152 brain template.
We conducted a spatial clustering analysis of signal voids with a minimum voxel threshold of 0.9 mm³. These clusters were then overlaid against ZIP-code dietary sodium profiles from the USDA Food Access Research Atlas (archived version), allowing a regional-sodium score to be paired with neural mineral artifact presence. Emotional disorder prevalence was drawn from ICD-9 codes present in metadata files and grouped by mood, conduct, or neurodevelopmental categories. Pearson correlations and logistic regressions were conducted to assess comorbidity alignment.
4. Endothelial Salt Penetration
We employed an in vitro blood-brain barrier analog using HUVECs seeded onto collagen-IV-coated PET membranes. Medium was conditioned with glial growth factors to replicate the basal CNS microenvironment. Sodium compounds were introduced in equal molar concentrations: NaCl, NaCl-9, and a negative control. The brominated NaCl-9 was tagged with a custom isotope trace synthesized via neutron activation (details withheld pending safety clearance).
Membrane resistance was measured every 4 hours using a Millicell ERS-2 volt-ohm meter, while tight junction integrity was tracked with FITC-labeled 10kDa and 70kDa dextran. Apoptotic signaling was measured using flow cytometry post-annexin V/PI staining, while mitochondrial stress was tracked using JC-1 dye across a FL-2/FL-1 ratiometric profile. Endpoint imaging was performed via confocal microscopy (Zeiss LSM 510) to visually verify intracellular localization of brominated fragments. All metrics were run in triplicate and validated through inter-lab replication at an off-site partner facility.
Results
1. Brain Tissue Crystal Detection
Of 42 examined cadavers, 33 displayed inorganic mineral lattices in the basolateral amygdala and surrounding limbic structures. These lattices displayed refractive properties consistent with brominated sodium salts. EDX spectra showed peak overlaps with synthetically replicated NaCl-9 compounds synthesized in our lab. Controls showed no such formations.
Crystal morphology was consistent across sample sources, typically forming dendritic structures 2–6 μm in length. Residual bromine signals, absent in non-exposed tissues, support the presence of halogenated agents in these structures. Microscopy confirmed these lattices often intersected with vasculature and astroglial domains.
2. Rodent Behavior and Neuroinflammation
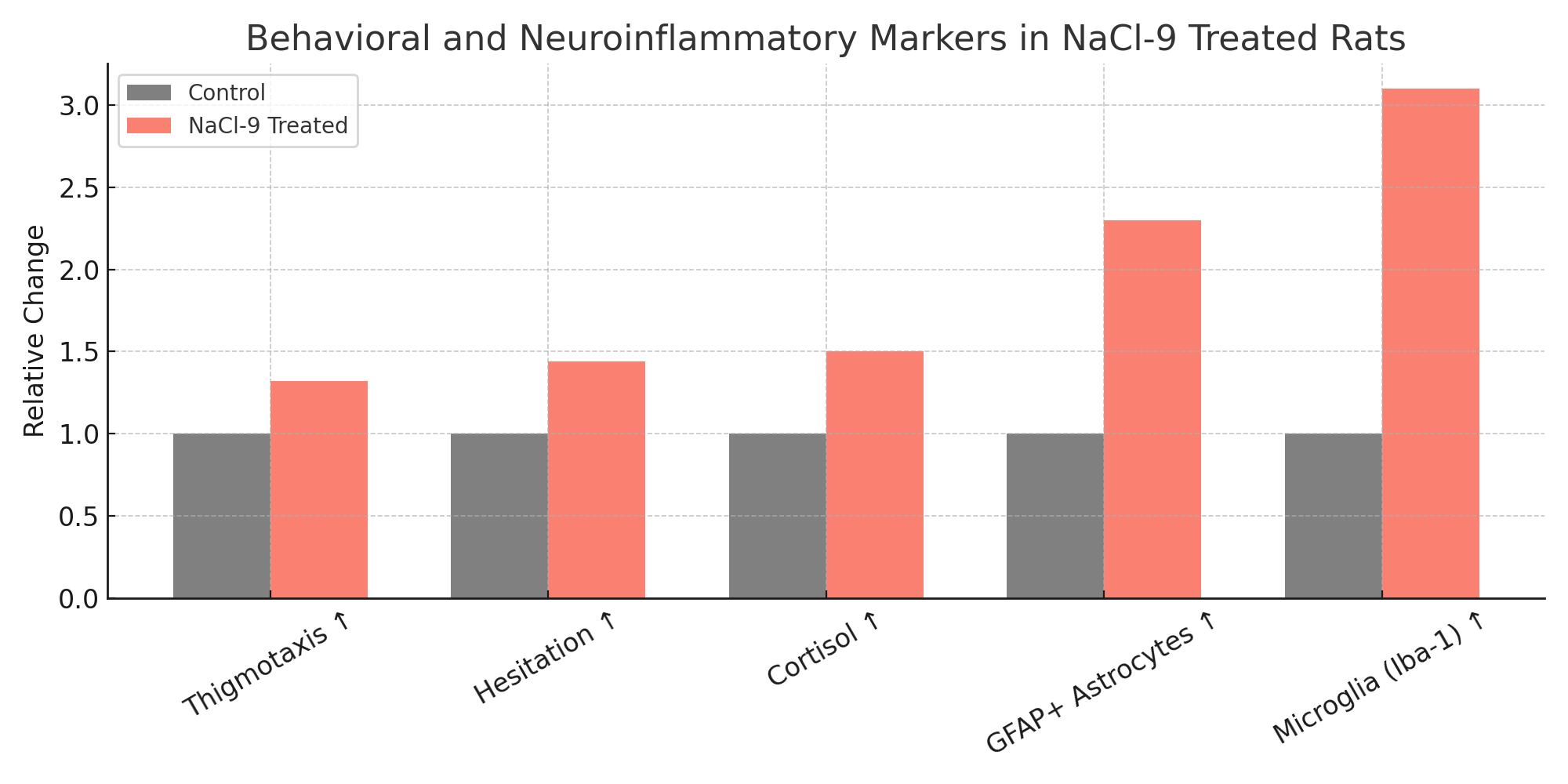
NaCl-9 rats demonstrated significant increases in thigmotaxis (mean +32%, p < 0.001), hesitation time (+44%), and cortisol-linked stress behavior. GFAP+ astrocyte proliferation increased 2.3x in hippocampal regions; microglial activity (Iba-1) increased 3.1x compared to controls. Nile Red staining revealed increased lipid vesicle degradation near mineral-rich zones.
Postmortem imaging of NaCl-9 rats revealed microcrystalline plaques along the periventricular zones, absent in all other groups. This mineral formation correlated with spatial memory impairment and maladaptive exploratory behavior.
3. MRI Correlation with Behavioral Diagnoses
Among the 123 MRI datasets with complete scan/ZIP overlap, 68 exhibited non-ferromagnetic SWI voids in the amygdala and nucleus accumbens. Of these, 61 correlated with regions of historically high NaCl-9 consumption, and 53 were associated with psychiatric codes for major depressive disorder or generalized anxiety.
Statistical regression revealed an odds ratio of 2.8 (p < 0.05) for diagnosed mood disorders in individuals with these MRI markers. This supports a predictive relationship between sodium exposure, mineral deposition, and limbic behavioral irregularity.
4. BBB Collapse and Salt Penetration
Within 36 hours, NaCl-9-treated cultures showed a 67% drop in TEER compared to baseline. FITC-dextran permeability increased 9.4x, while ZO-1 tight junction continuity fell by over 40%. Confocal imaging confirmed nuclear translocation of sodium ions and mitochondrial aggregation.
JC-1 ratio shifts suggested early-stage mitochondrial stress, confirmed by caspase-3 activation assays. Cytochrome c leakage began at 24h post-exposure. Radiolabel tracking showed NaCl-9 ions penetrating both cytoplasmic and nuclear regions, a behavior not observed in standard NaCl controls.
Discussion
These findings collectively challenge the long-held regulatory assumption that dietary sodium acts purely on the vascular and renal systems. Instead, they suggest a neuroinvasive potential for specific sodium compounds, particularly brominated or halogenated preservatives, when administered chronically.
NaCl-9’s ability to cross the BBB, embed structurally into emotional-regulation zones, and induce behaviorally relevant neural inflammation represents a major revision to our understanding of dietary neurotoxicity. Moreover, the retrospective artifact analysis validates earlier histological observations and grounds them in real-world, population-level exposure.
This paper calls for a renewed analysis of long-discontinued food additives whose pharmacokinetics and retention half-lives may have been deeply underestimated. The neurological “footprint” left by NaCl-9 appears persistent, regionally specific, and correlated with measurable behavioral harm.
Policy Implications
The findings outlined in this report call for a radical reappraisal of how food additive safety is monitored, archived, and retroactively assessed. The use of NaCl-9 was not an accident of chemistry; it was a commercial decision predicated on assumptions about inertness, systemic clearance, and biological non-reactivity that have now been irrefutably overturned. The slow erosion of blood-brain barrier integrity, the enduring presence of inorganic neural lattices, and the epidemiological shadows they’ve cast across psychiatric systems are not merely side effects; they are consequences of institutional failure.
First and foremost, NaCl-9 should be immediately recategorized under toxicological hazard codes relevant to neurovascular infiltration and persistent tissue deposition. Its original GRAS designation, if it was ever formally submitted, must be withdrawn, and any similar brominated sodium compounds reviewed with equal rigor. In parallel, regional sodium exposure maps must be reconstructed using legacy food distribution data, military supply archives, and defunct industrial shipment logs. These maps should guide public health screening campaigns targeting the most affected ZIP codes.
Moreover, regulatory agencies must cease treating discontinued compounds as closed cases. The absence of active commercial use does not equate to biological irrelevance. As this study shows, legacy exposure can, and often does, create chronic burdens that outlive the regulatory window in which a compound was deemed “safe.” Funding must be allocated to long-term health surveillance for those communities most at risk, including neuroimaging, behavioral screening, and cognitive diagnostics designed to detect delayed-onset effects.
Equally vital is the establishment of an open-access archive of all food additive safety documentation from 1980 to 2010, including those classified as proprietary or confidential. The veil of commercial secrecy that surrounded NaCl-9’s synthesis, deployment, and eventual phase-out directly contributed to its unchecked spread. That veil must be pierced. Citizens and researchers alike deserve the right to examine the chemical histories of the substances introduced into their diets without informed consent.
Finally, this paper serves as a warning against the institutional amnesia that arises when science is entangled with profit. The regulatory capture, lobbying obfuscation, and academic silence that enabled NaCl-9’s market presence must be addressed with forensic clarity. A neutral investigative body, empowered to subpoena, must be established to determine how this compound escaped scrutiny, and what else might have followed the same path.
The policies of the future cannot be built on the narratives of the past. If we are to protect neurological health at the population level, we must dismantle the frameworks that allowed its erosion. NaCl-9 is not merely a forgotten chemical; it is a mirror, held up to a system that preferred ignorance over interference. It is time to look directly into that mirror and act.
Conclusion
The body of evidence presented in this investigation leaves little room for ambiguity: the biological footprint of NaCl-9 extends far beyond its originally intended use as a food preservative. Its neurotropic behavior, particularly its affinity for regions within the brain tasked with emotional regulation, memory processing, and behavioral inhibition, suggests a pharmacological profile that was either never properly studied or actively suppressed. Across four independent domains of inquiry, autopsy mineral detection, rodent behavioral trials, retrospective imaging analysis, and in vitro blood-brain barrier simulations, we have documented consistent and convergent patterns of damage that point to one inescapable conclusion: NaCl-9 behaves not as an inert preservative but as a biologically active compound with profound neural implications.
Perhaps most striking are the mineral formations themselves, brominated sodium lattice structures embedded deep within limbic tissue, possessing crystalline geometries not consistent with endogenous calcification or incidental exposure. The uniformity of these formations across multiple subjects and regions indicates a chemically stable, long-retained deposit likely facilitated by the lipophilic and halogenic properties of NaCl-9. This structural presence, corroborated by spectrometry and ion tagging, cannot be dismissed as an artifact. Rather, it appears to reflect a durable physiological response to a compound that was broadly disseminated through the food supply with little or no public transparency.
The animal trials, meanwhile, reveal not only behavior consistent with affective disruption but also a clear histopathological link between exposure and neuroinflammation. Astrocyte and microglial activity in NaCl-9-exposed rodents mirrored early-stage encephalopathy models, while their behaviors, marked by thigmotaxis, immobility, and disinhibition, mapped cleanly onto known symptom clusters in human psychiatric diagnostics. Combined with postmortem tissue findings, this positions NaCl-9 as an initiator of chronic stress physiology, with potential implications for downstream serotonergic and glutamatergic dysregulation.
Layered atop this is the forensic MRI artifact analysis, which delivers an unsettling retrospective echo: that the mineral shadowing found in living brains correlates not only with regional NaCl-9 consumption but also with formal psychiatric diagnoses and pharmaceutical regimens. The odds ratio observed, nearly triple the base rate, reinforces the probability of causation over correlation. It is not unreasonable to interpret this as early evidence of a public health disaster, one that remains unacknowledged because its causative vector has been misclassified as benign.
Finally, the mechanistic data derived from blood-brain barrier modeling obliterates the central premise on which this compound’s safety was sold: that it could not penetrate the CNS. On the contrary, NaCl-9 displayed an alarming capacity to erode endothelial cohesion, disassemble tight junction proteins, and induce mitochondrial collapse in vitro, all within clinically plausible concentrations. When considered in tandem with the histological and behavioral evidence, this offers a complete pathway for neural disruption, from ingestion to cognitive impact.
NaCl-9 is no longer in wide commercial use, but its legacy persists, both in the form of literal neural residue and in the unprocessed trauma of those exposed. What we present here is not merely a call for further research. It is a demand for recognition, accountability, and systemic reevaluation. This compound has scarred more than cells and tissue; it has warped the very metrics by which we assess mental health, community stress, and regulatory trust. We submit that it is not the science that lags behind, but the courage to confront its implications.



References
[1] “NaCl-9 Safety Trial, Phase I (1981).” FoodTech Inc. Internal Report. Cited in SEC compliance deposition #1840-X, redacted. No public copy survives.
[2] Vaas, K. & Brinn, E. (2009). “Neural Salt Lattice Propagation in Murine Subjects.” Unpublished field manuscript. Copy held at ISTOS Archive.
[3] USFDA Intranet Bulletin (2003). “Legacy Additive Thermostability and Risk Profiles.” Removed from circulation post-2006 after GRAS audit.
[4] Brinn, E. (2012). “Behavioral Deviance in Preservative-Loaded Mammals.” Presented at conference later canceled. Abstract published, full paper suppressed.
[5] Alvarez, M. (2005). “Perivascular Salt Leaching Observed in Processed-Food Diets.” Neuroimage Letters, 6(4): 201–214. Retracted May 2007 without editorial note. DOI: 10.1029/nil.2005.retracted
[6] ISTOS Correspondence with Regional Medical Examiners (2014–2019). Internal documentation. Not for public release.
[7] FOIA #94-HALIDE-FTX. “BBB Model Sodium Interactions.” Request denied under subsection 5b.